9 applications of machine learning in drug discovery
Top ways you can use machine learning to transform drug discovery and development
10 min read
June 28th, 2023
Last updated: July 23rd, 2024


Machine learning has revolutionized the field of drug discovery, providing new and innovative approaches to identify potential drug candidates, predict toxicity, optimize chemical reactions, design multi-target drugs, etc.
In this blog post, we will explore different applications of machine learning in drug discovery.
We will discuss the latest research and developments in each of these areas, as well as the potential impact that machine learning can have on the drug discovery process.
Whether you are a researcher in the field of drug discovery or simply interested in the latest advances in machine learning, this blog post is for you.
Potential drug candidate identification
Machine learning has been widely used in the identification of potential drug candidates due to its ability to quickly analyze and extract valuable information from large datasets.
In this section, we will explore some of the recent studies that have utilized machine learning in the identification of potential drug candidates.
The researchers in a study built machine-learning predictive models to identify new drug candidates that can target the viral proteins 3 chymotrypsin-like protease (3CLpro) and RNA-dependent RNA polymerase (RdRp).
They collected curated training sets of substances from CAS data collections and integrated them with curated bioassay data. The best-performing classification models were then used to screen a set of FDA-approved drugs and CAS REGISTRY substances with antiviral properties.
Several potential substances that could target 3CLpro or RdRp were identified, and some were validated by previous bioassay studies or included in ongoing COVID-19 clinical trials. The study demonstrates that machine learning-based predictive models can be useful in the drug discovery process for COVID-19 and other diseases.
In another study, researchers developed a machine learning approach combining generative modeling and reinforcement learning to address the challenges in identifying potent compounds for Parkinson's disease.
Training data from an assay measuring inhibition of secondary nucleation were used to train the model. The method successfully identified small molecules with high potency against secondary nucleation, offering potential avenues for the development of effective treatments for Parkinson's disease.
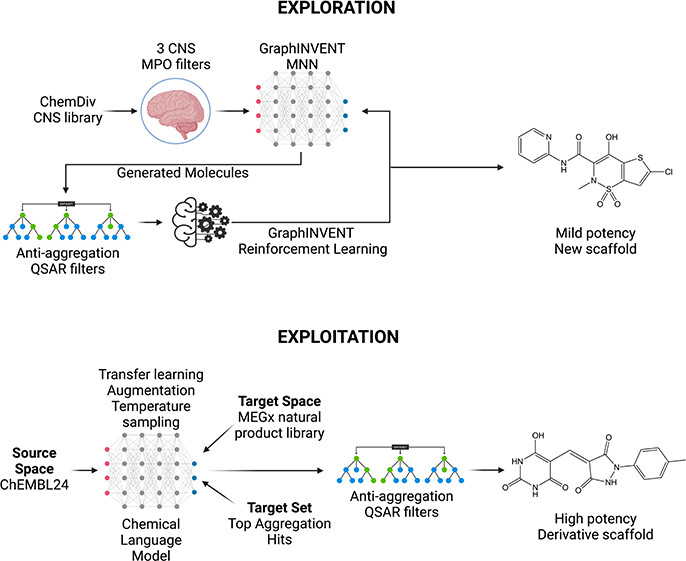
Image Source: J. Chem. Theory Comput.
Machine learning has also been utilized in the identification of potential drug candidates for cancer.
In one study, researchers aimed to discover new drugs with antisarcoma activity using large datasets from multiple preclinical assays with diverse experimental conditions.
They utilized the ChEMBL database, which provided a comprehensive collection of antisarcoma assay outcomes and chemical compounds.
In this work, the researchers proposed a PTML (perturbation theory and machine learning) model that integrated linear discriminant analysis and neural network approaches.
Machine learning has shown great promise in the identification of potential drug candidates by analyzing large datasets and predicting the pharmacokinetic and pharmacodynamic properties of compounds. By leveraging the power of machine learning, you can also accelerate the drug discovery process and identify novel compounds with therapeutic potential.
Check out some powerful curricula available on Neovarsity to learn machine learning and artificial intelligence for drug discovery. These programs harness the potential of machine learning to accelerate the drug discovery process and identify novel compounds with therapeutic potential.
Discover your potential in data-driven drug discovery!
Get started in machine learning for drug discovery with confidence! This course provides a comprehensive introduction to core concepts and essential techniques in molecular machine learning.
- Learn essential techniques in molecular machine learning
- Master molecular ML model building and quality control for drug discovery
- Apply skills to small molecule tasks, including virtual screening
Toxicity prediction
Toxicity prediction is a critical step in drug discovery and chemical risk assessment as it allows researchers to identify potentially toxic compounds and avoid their development or use.
Machine learning has emerged as a powerful tool for predicting the toxicity of chemicals and drugs.
Let us explore some recent studies that have utilized machine learning in the prediction of toxicity.
In one study, researchers introduced a high-throughput virtual screening tool called "ED Profiler". This tool incorporates quantitative structure-activity relationship (QSAR) models for nonreceptor-mediated targets, such as human and fish hormone transport proteins.
The QSAR model was developed by utilizing machine learning algorithms like k-nearest neighbor (kNN) or decision tree. It was implemented in Python programming language and enables the prediction of potential disrupting effects of endocrine-disrupting compounds (EDCs) on nonreceptor-mediated targets.
In another study, researchers focused on the in silico prediction of chemical-induced hematotoxicity using machine learning and deep learning methods.
They compiled a comprehensive dataset comprising 632 hematotoxic chemicals and 1525 approved drugs without hematotoxicity. The researchers employed various machine learning and deep learning algorithms integrated within the Online Chemical Modeling Environment (OCHEM) to develop computational models.
By leveraging the three best individual models, a consensus model was constructed, achieving a prediction accuracy of 0.83 and a balanced accuracy of 0.77 on external validation. The consensus model, along with the best individual model developed using the random forest regression and classification algorithm (RFR) and QNPR descriptors, can be accessed at https://ochem.eu/article/135149.
In another study, researchers aimed to develop a QSAR model for predicting the comedogenic potential of cosmetic ingredients.
The study utilized various machine learning algorithms and types of molecular descriptors to build the model.
The objective was to enhance the understanding and prediction of the comedogenicity of cosmetic ingredients, contributing to the development of safer and more effective cosmetic products.
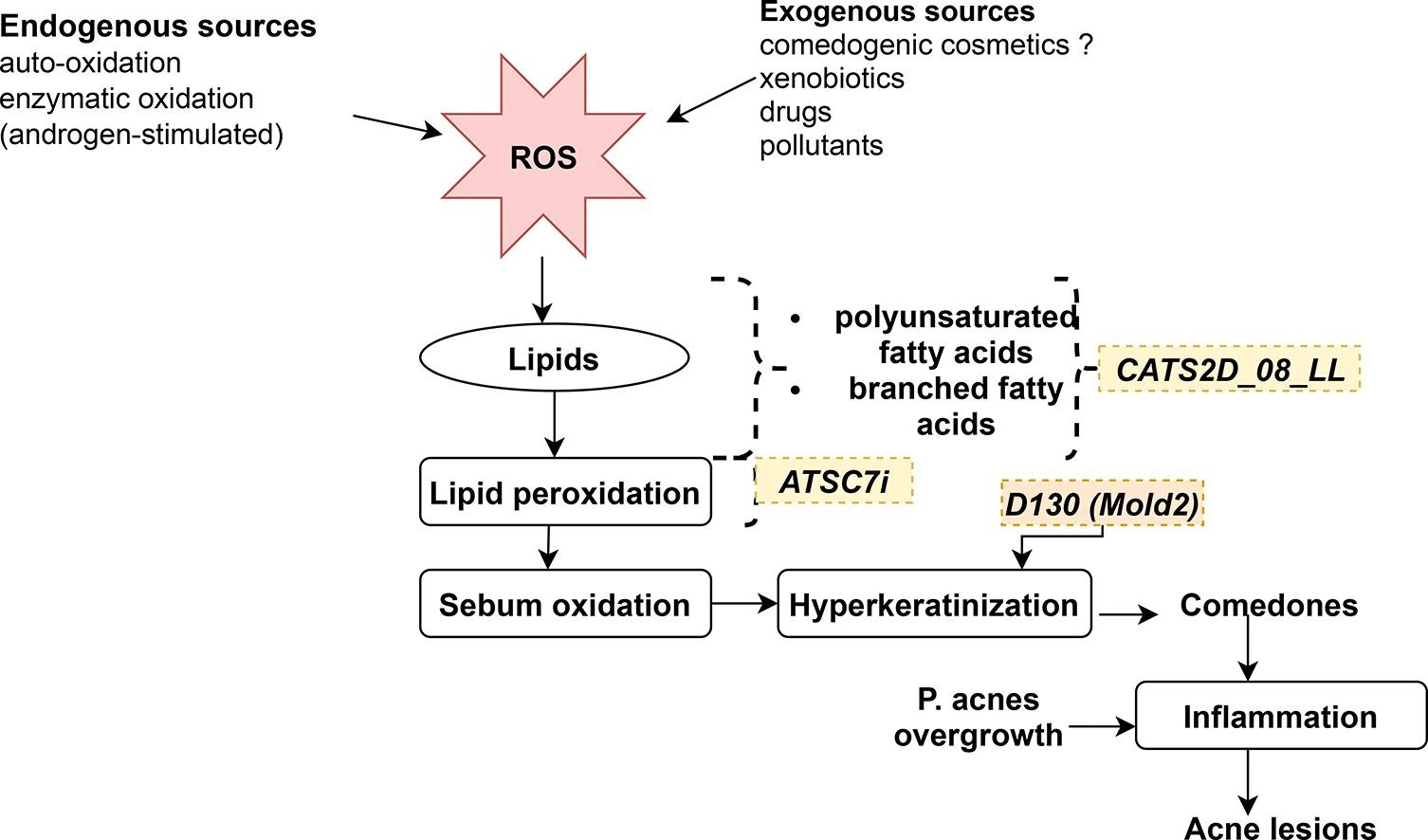 Image Source: Computational Toxicology
Image Source: Computational Toxicology
Virtual screening of large databases
Virtual screening of large databases has become a key step in drug discovery, enabling researchers to identify potential drug candidates rapidly.
Machine learning has emerged as a powerful tool for virtual screening due to its ability to analyze large datasets and make accurate predictions.
Let us explore recent studies that have utilized machine learning in the virtual screening of large databases.
In one study, researchers introduced a novel machine learning framework called MEMES (Machine learning framework for Enhanced MolEcular Screening) based on Bayesian optimization. This framework aims to efficiently sample the chemical space for enhanced molecular screening.
The study demonstrated that MEMES successfully identified 90% of the top 1000 molecules from a vast molecular library comprising approximately 100 million compounds.
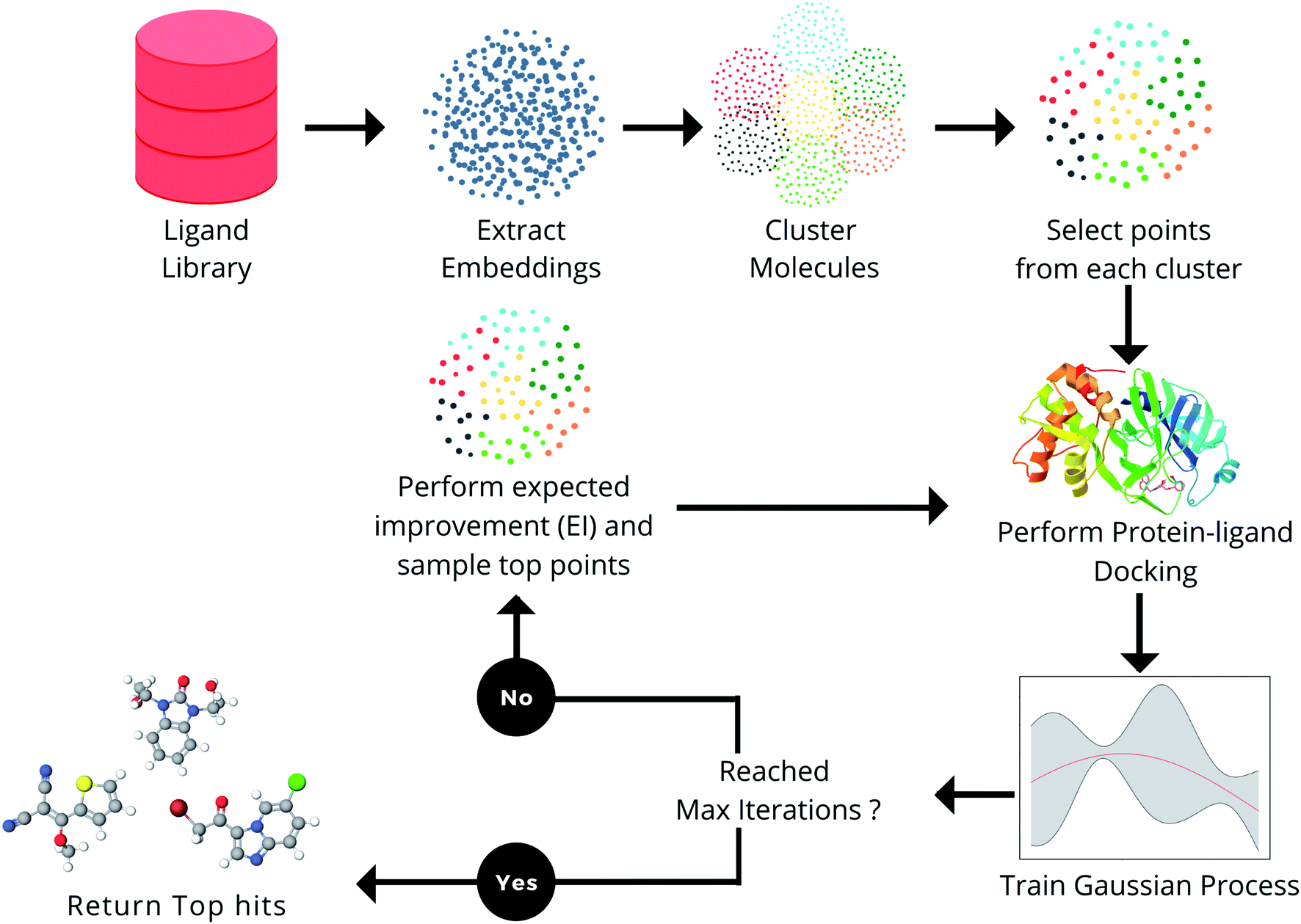
Image Source: Chemical Science
Remarkably, the framework achieved this outcome while only calculating the docking score for approximately 6% of the complete library. The researchers anticipate that such a framework will significantly reduce computational efforts in drug discovery and other fields that require high-throughput experiments.
In another study, researchers introduced a machine learning-enabled pipeline for large-scale virtual screening.
The pipeline involved clustering compounds based on molecular properties, followed by limited docking against a specific drug target. The library was subsequently trimmed by a factor of 10, and the remaining compounds were individually screened using docking techniques.
Finally, a dense neural network was trained to classify the identified hits into true and false positives. As a demonstration, the researchers applied this pipeline to screen for inhibitors against RPN11, the deubiquitinase subunit of the proteasome, which is a drug target for breast cancer.
Target identification
Target identification is a critical step in drug discovery, as it enables researchers to identify proteins or other molecules that can be targeted by potential drugs. Machine learning has emerged as a powerful tool for target identification, as it can analyze large datasets and identify patterns that are not easily discernible through traditional methods.
In one study, researchers introduce BANDIT, a Bayesian machine-learning approach that leverages multiple data types to predict drug binding targets.
By integrating public data, BANDIT achieved an impressive accuracy of approximately 90% on over 2000 small molecules. When applied to more than 14,000 compounds lacking known targets, BANDIT successfully generated around 4,000 novel molecule-target predictions.
Among these predictions, 14 novel microtubule inhibitors were validated, including three that exhibited activity against resistant cancer cells. The researchers also applied BANDIT to ONC201, an anti-cancer compound with an elusive target.
Through BANDIT, they identified and validated DRD2 as the target for ONC201, leading to a more precise clinical trial design.
Additionally, BANDIT revealed connections between different drug classes, shedding light on previously unexplained clinical observations and presenting new opportunities for drug repositioning.
In another study, researchers focused on the identification of potential targets for known bioactive compounds, a critical aspect of drug design and development.
They employed molecular docking, a computational approach where a molecule is docked into various protein structures to predict potential targets. However, the accuracy of reverse docking approaches is limited by the scoring functions' ability to distinguish between targets and nontargets.
To address this limitation, the researchers developed target-specific scoring functions using supervised machine learning techniques, specifically neural networks and support vector machines (SVMs).
These scoring functions, trained on bioactivity data obtained from ChEMBL, demonstrated improved prediction performance for target identification across multiple validation datasets.
The researchers utilized protein-ligand interaction fingerprints derived from docking poses of active and inactive compounds as input for training the models.
The developed models were evaluated on previously unseen molecules, and the results showcased their efficacy in correctly ranking and predicting targets for both single-target selectivity and multitarget datasets.
This study highlights the potential of target-specific scoring functions combined with molecular docking to enhance the accuracy and efficiency of in-silico protein target prediction for drug discovery.
Machine learning has also been used in the prediction of protein-protein interactions, which can help identify potential drug targets.
In one study, researchers have developed a machine learning model to predict protein-protein interface hotspots by integrating multiple features, including those related to amino-acid chains.
The model's effectiveness has been evaluated, demonstrating improved accuracy in identifying these hotspots. Furthermore, the researchers conducted virtual drug screening analysis on a set of hotspots identified in the EphB2-ephrinB2 complex.
The model exhibited promising predictive capabilities, with an AUROC (area under the receiver operating characteristic curve) of 0.842, sensitivity/recall of 0.833, and specificity of 0.850.
These findings highlight the potential of this machine learning model for enhancing the identification of protein-protein interface hotspots and supporting virtual drug screening efforts.
Binding affinity prediction
Binding affinity prediction is an important task in drug discovery that aims to predict the strength of the binding between a drug candidate and its target protein.
Accurately predicting binding affinity can help researchers select the most promising drug candidates and reduce the cost and time required for drug development.
Machine learning has been increasingly used in binding affinity prediction due to its ability to analyze large datasets and capture complex relationships between molecules and proteins.
Let us review recent studies that have utilized machine learning in binding affinity prediction.
In one study, researchers utilized SFCscore descriptors to enhance the scoring function used in drug discovery.
By employing a PDBbind training set of 1005 complexes and employing random forest for regression, they developed a new scoring function known as SFCscoreRF.
The performance of SFCscoreRF was found to be significantly improved compared to previously developed SFCscore functions, as demonstrated on the PDBbind and CSAR-NRC HiQ benchmarks.
However, leave-cluster-out cross-validation and evaluation in the CSAR 2012 scoring exercise revealed some limitations and indicated areas for further improvements in SFCscoreRF and empirical scoring functions in general.
In another study, researchers aimed to enhance the prediction accuracy of protein-ligand complex binding affinity by proposing a deep-neural network model.
The model incorporated two crucial features: descriptor embeddings, which provided information on the local structures of the protein-ligand complex, and an attention mechanism to emphasize important descriptors for binding affinity prediction.
The performance of the proposed model surpassed that of existing binding affinity prediction models on various benchmark datasets. The study confirmed the effectiveness of the attention mechanism in capturing binding sites within protein-ligand complexes, ultimately improving prediction performance.
The code for the proposed model is publicly available here.
Become a machine learning expert in drug discovery!
This is an end-to-end curriculum, which also teaches advanced concepts in explainable and interpretable machine-learning techniques suited for building reliable models for drug discovery applications.
- Implement machine-learning algorithms in diverse drug discovery applications
- Master advanced topics in molecular data analytics
- Handle biases in molecular data modeling effectively
- Showcase your expertise through practical capstone projects
Chemical reaction optimization
Chemical reaction optimization is a critical step in the drug discovery process, where researchers seek to optimize the chemical reaction conditions to maximize product yield while minimizing unwanted side reactions.
Machine learning has emerged as a promising tool to assist in chemical reaction optimization, by enabling researchers to quickly and accurately identify the optimal reaction conditions.
In one study, researchers generated an open-source dataset comprising more than 30,000 organic chemistry gas phase partition functions.
Using this dataset, they trained a machine learning deep neural network estimator capable of predicting partition functions for unknown organic chemistry gas phase transition states.
The estimator relies solely on reactant and product geometries along with partition functions. Additionally, a second deep neural network was trained to predict partition functions of chemical species based on their geometry.
The models demonstrated accurate predictions of the logarithm of partition functions in the test set, with a maximum mean absolute error of 2.7%. This approach offers a cost-effective means to compute reaction rate constants ab initio.
Moreover, the models successfully computed transition state theory reaction rate constant prefactors, exhibiting quantitative agreement with corresponding ab initio calculations at an accuracy of 98.3% on the logarithmic scale.
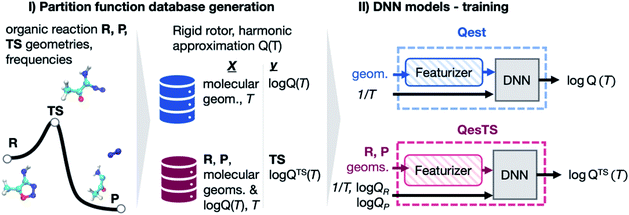
Image Source: Chemical Science
In another study, researchers have developed a novel approach combining a data-driven machine learning model with reactivity descriptors to predict optimal enzyme-catalyzed synthesis conditions and reaction yields.
A total of fourteen reactivity descriptors were constructed to represent 125 reactions across various reaction mechanisms and categories. Nineteen machine learning models were trained using the dataset, with the Quadratic support vector machine (SVM) model demonstrating the best performance.
This model was then employed to predict the optimal reaction conditions, enabling the attainment of the highest yield among 109,200 different reaction conditions involving variations in substrates, solvents, water contents, enzyme concentrations, and temperatures. The proposed protocol offers a versatile solution applicable to diverse chemical reactions, providing a black-box evaluation for optimizing reaction conditions in organic synthesis reactions.
Binding sites prediction
In the field of drug discovery, identifying the binding sites of protein targets is crucial for the design of small-molecule therapeutics.
Machine learning algorithms have shown great potential for predicting the binding sites of proteins with high accuracy.
In one study, researchers introduced P2Rank, a machine learning-based tool for ligand binding site prediction. P2Rank utilizes the prediction of ligandability in local chemical neighborhoods centered on points placed on the protein's solvent-accessible surface.
The study demonstrated that P2Rank outperformed existing tools such as Fpocket, SiteHound, MetaPocket 2.0, and DeepSite in terms of performance. Additionally, P2Rank offers fast prediction times (under 1 second per protein) and a multi-threaded implementation, making it suitable for large datasets and scalable structural bioinformatics pipelines.
P2Rank is available as a user-friendly stand-alone command line program and a Java library, requiring minimal dependencies and enabling fully automated predictions.
In another study, researchers focused on developing a system capable of predicting the binding sites of proteins for five mononucleotides: AMP, ADP, ATP, GDP, and GTP.
The system consisted of two machine learning-based predictors utilizing a convolutional neural network and a gradient boosting machine, along with two template-based predictors relying on sequence and structure alignment.
Additionally, an ensemble learning predictor was employed, combining the outputs of the four predictors. To enhance the performance of the system, data augmentation techniques were applied by including ligand binding sites with similar ligand structures.
This study presents a comprehensive approach for accurately predicting protein binding sites for specific mononucleotides.
Drug repurposing
Drug repurposing, or the identification of new therapeutic uses for existing drugs, has become an attractive approach to expedite drug development and reduce costs.
Machine learning has shown great potential in the field of drug repurposing, enabling researchers to efficiently identify new therapeutic uses for existing drugs.
In one study, researchers introduce DRIAD (Drug Repurposing In AD), a machine learning framework aimed at quantifying potential associations between the severity of Alzheimer's disease (AD) pathology (measured by the Braak stage) and molecular mechanisms represented by lists of gene names.
DRIAD was applied to gene lists derived from perturbations in differentiated human neural cell cultures caused by 80 FDA-approved and clinically tested drugs.
The framework generated a ranked list of potential drug repurposing candidates. The top-scoring drugs were further analyzed to identify common trends among their targets.
The researchers propose that the DRIAD method could serve as a means to nominate drugs for further evaluation in clinical trials, provided additional validation and identification of relevant pharmacodynamic biomarkers are conducted.
In another study, researchers propose a novel approach for drug repurposing using a two-stage prediction and machine learning framework.
The first stage involves clustering diseases based on gene expression patterns to identify shared critical pathways across different disease conditions.
In the second stage, drug efficacy is evaluated by assessing the reversibility of abnormal gene expression and clustering the results to identify potential repurposing targets.
To cluster diseases, gene expression data from 262 cases of 31 diseases and 268 controls were analyzed using Uniform Manifold Approximation and Projection for Dimension Reduction, followed by k-means clustering.
Disease-specific gene expression data for inclusion body myositis, polymyositis, and dermatomyositis were examined, and the LINCS L1000 characteristic direction signatures search engine (L1000CDS2) was utilized to identify small-molecule compounds that reversed the expression patterns of specifically altered genes, serving as repurposing candidates.
The functions of affected genes were further analyzed using Gene Set Enrichment Analysis to assess consistency with expected drug efficacy.
Through this approach, the researchers identified 22 potential repurposing candidates, including KM 00927, I-BET, alvocidib, and vorinostat.
Discover your potential in data-driven drug discovery!
Get started in machine learning for drug discovery with confidence! This course provides a comprehensive introduction to core concepts and essential techniques in molecular machine learning.
- Learn essential techniques in molecular machine learning
- Master molecular ML model building and quality control for drug discovery
- Apply skills to small molecule tasks, including virtual screening
Multi-target drug design
Multi-target drug design (MTDD) involves the identification of compounds that interact with multiple targets, providing a more effective treatment option for complex diseases.
Machine learning has shown great potential in MTDD, enabling researchers to efficiently identify compounds that have interactions with multiple targets. In this report, we will review recent studies that have utilized machine learning in MTDD.
In one study, researchers introduce multi-target-based polypharmacology prediction (mTPP) as an approach for exploring the relationship using virtual screening and machine learning techniques. The study focuses on predicting the activity of potential hepatoprotective components.
To construct the mTPP model, the researchers utilize data on the binding strength of individual ingredients with multiple targets and the proliferation rate of compounds against acetaminophen (APAP)-induced injury in L02 cells.
The mTPP model is constructed using algorithms such as Multi-layer Perceptron (MLP), Support Vector Regression (SVR), Decision Tree Regressor (DTR), and Gradient Boost Regression (GBR).
In another study, researchers delved into the potential of explainable machine learning in identifying characteristic structural motifs of dual-target compounds.
They focused on a pharmacologically relevant target pair-based test system, developing accurate prediction models.
To gain insights into the predictions, the researchers quantified the influence of molecular representation features of test compounds and identified specific features that were present in dual-target compounds but absent in single-target compounds.
These features formed coherent substructures within the dual-target compounds. Through computational analysis of feature contributions, the researchers unveiled structural motifs that served as signatures of different dual-target activities.
This study demonstrates how explainable machine learning can bridge the gap between predictions and chemical analysis, providing valuable insights into the characteristic substructures of dual-target compounds.
Summary
In this comprehensive blog post, we explored the fascinating applications of machine learning in drug discovery.
We delved into various areas where machine learning has made significant contributions, including drug candidate identification, toxicity prediction, binding site prediction, target identification, binding affinity prediction, drug repurposing, virtual screening, chemical reaction optimization, and multi-target drug design.
We discussed the latest research and developments in each of these areas, highlighting the potential impact of machine learning on the drug discovery process.
To further explore and gain expertise in the exciting domain of machine learning and artificial intelligence for drug discovery and digital chemistry, consider enrolling in Neovarsity's specialized courses.