A Beginner's Guide to QSAR Modeling in Cheminformatics for Biopharma
Learn the basics, applications, and key concepts in this beginner-friendly guide.
20 min read
August 13th, 2024
Last updated: April 18th, 2025

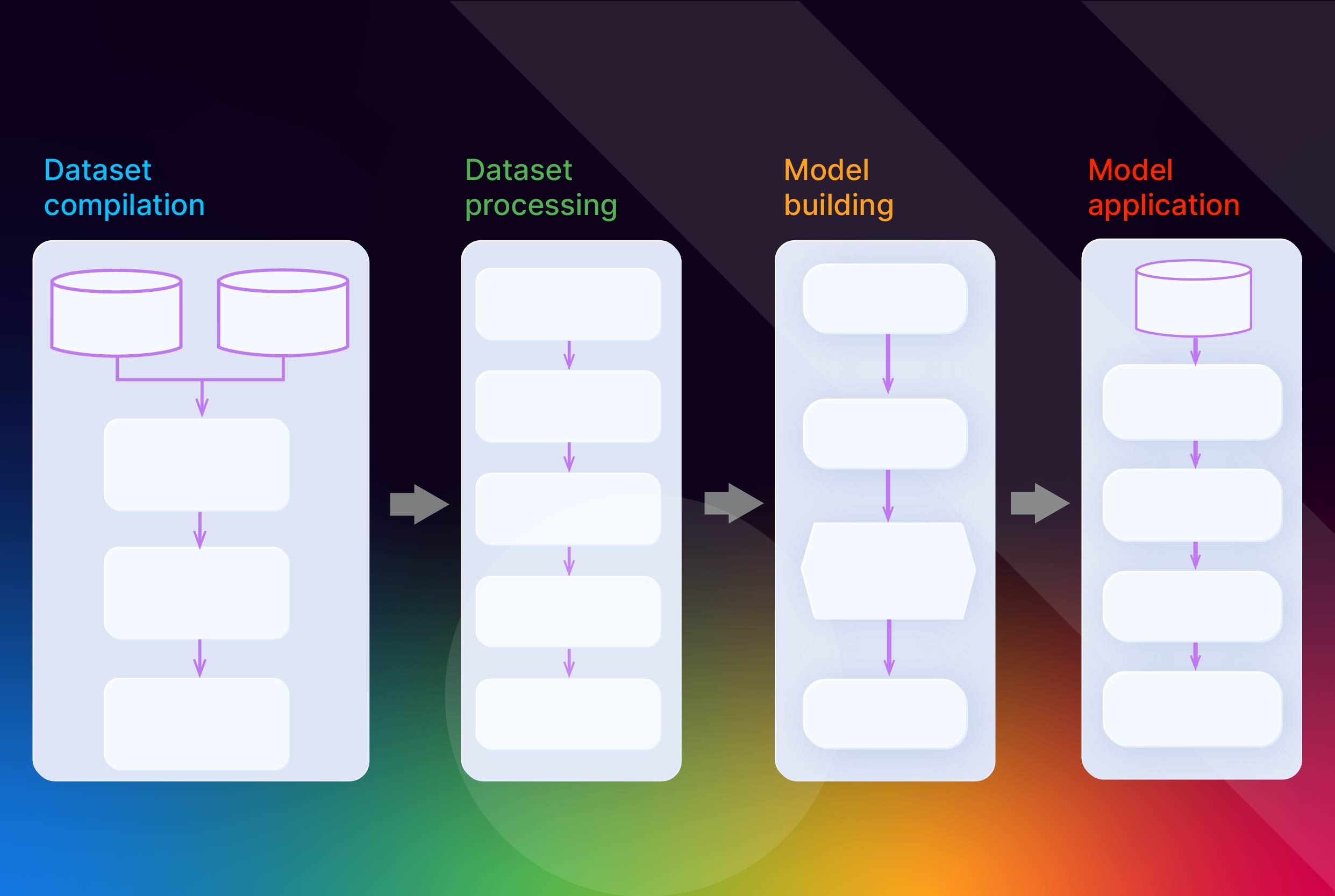
Introduction
Quantitative structure-activity relationship (QSAR) models mathematically link a chemical compound’s structure to its biological activity or properties. These models operate on the principle that structural variations influence biological activity. QSAR modeling uses physicochemical properties and molecular descriptors of chemicals as predictor variables, while biological activity or other chemical properties serve as response variables. By analyzing datasets of known compounds, QSAR models identify patterns between chemical structures and biological activity, enabling predictions for new compounds.
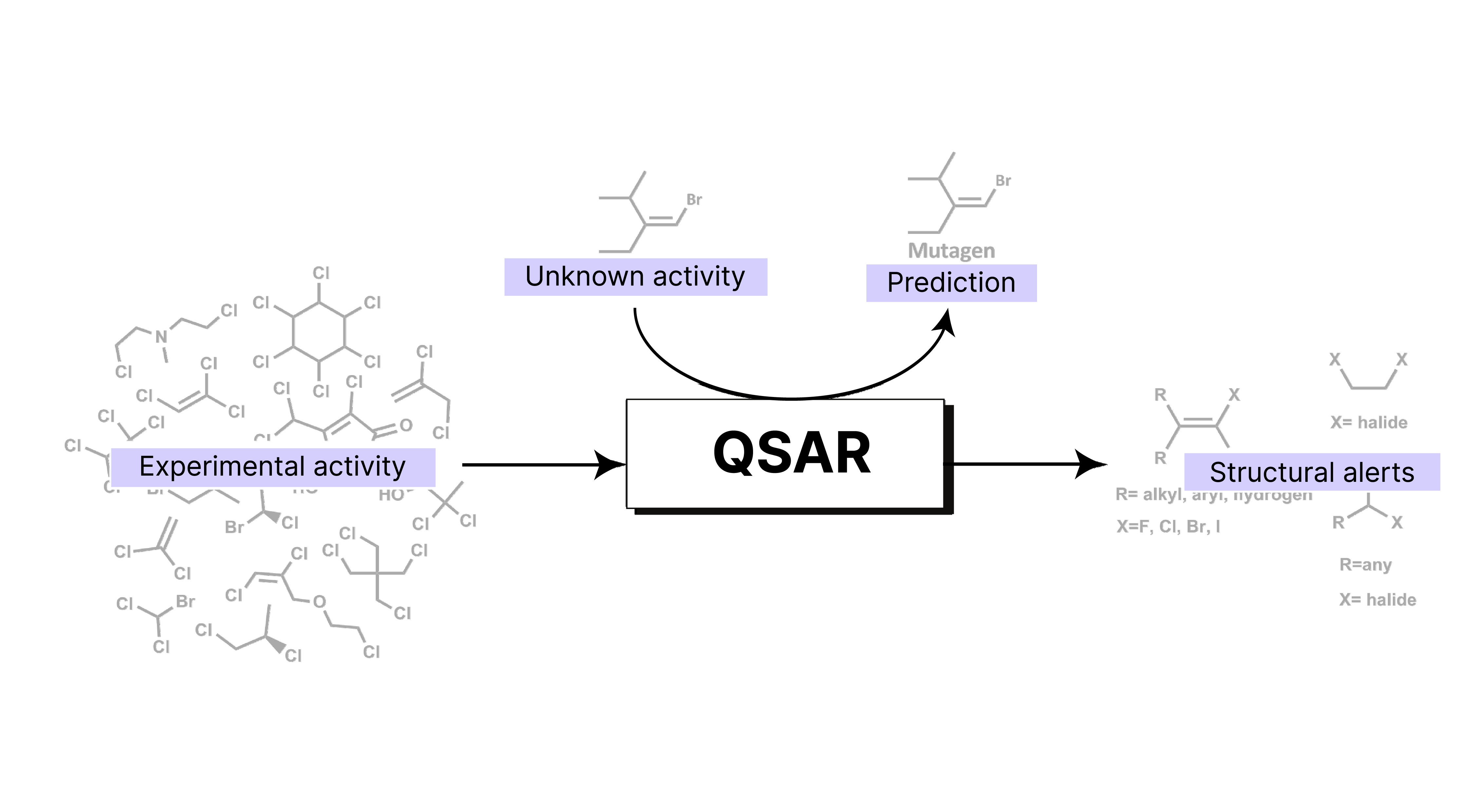
Image adapted from: Pérez-Sánchez Lab @ UCAM
What is QSAR modeling?
It is a computational approach used in cheminformatics to predict the biological activity or properties of chemical compounds based on their molecular structures.
QSAR modeling plays a crucial role in drug discovery and environmental chemistry by:
Prioritizing promising drugs: As an in-silico approach, QSAR modeling efficiently screens large compound libraries, prioritizing candidates with desired biological activity and minimizing the need for in vivo testing
Reducing animal testing: QSAR models serve as validated alternatives to animal testing and are increasingly adopted in regulatory frameworks for chemical risk assessment worldwide.
Predicting properties: QSAR models are used to predict the physicochemical, biological and environmental properties of compounds from the knowledge of their chemical structure.
Providing quantitative relationships: QSAR provides a mathematical model that quantitatively relates a numerical measure of chemical structure to a biological activity or property.
Guiding chemical modifications: Understanding the relationship between structure and activity can guide the design of new chemicals with improved properties.
Related Reading: 20 Must-Read Papers for a Strong QSAR Foundation in Drug Design
Fundamentals of QSAR
I. Basic Concepts of QSAR
Molecular Representations
QSAR models represent molecules as numerical vectors, with each element corresponding to a descriptor that quantifies structural, physicochemical, or electronic properties of the molecule. Common molecular descriptors include constitutional, topological, electronic, geometric and thermodynamic descriptors.
Structure - Activity Relationship
QSAR models aim to find a mathematical relationship between the molecular descriptors and the biological activity or property of interest, such as:
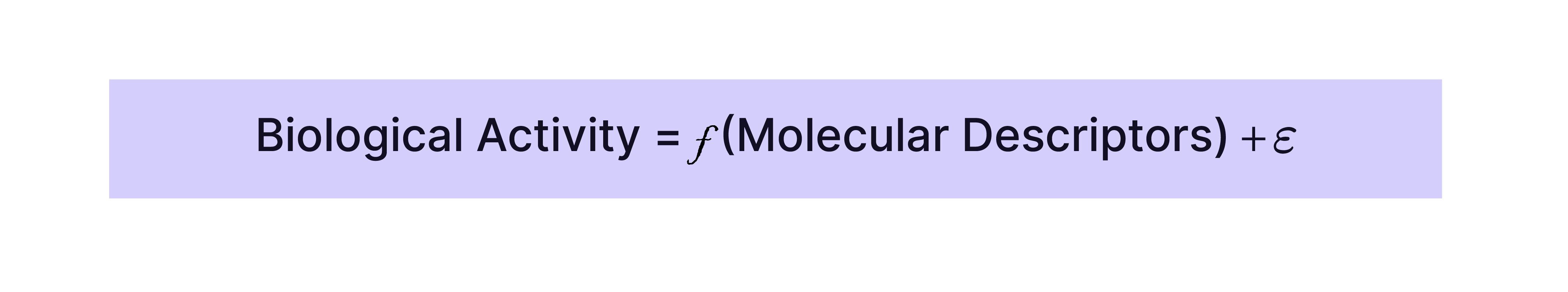
where ϵ represents the error or residual not explained by the model.
Model Development
The general workflow for developing a QSAR model involves:
- Curating a dataset of molecules with known biological activities.
- Calculating molecular descriptors for the dataset.
- Selecting the most relevant descriptors using variable selection techniques.
- Building a predictive model relating the descriptors to the activity using regression or classification methods.
- Validating the model's predictive performance using internal and external test sets.
Model Interpretation
QSAR models can provide insights into the structural features and physicochemical properties that influence biological activity. This can guide the design of new, more potent molecules. Common techniques for model interpretation include:
- Identifying the most important descriptors in the model.
- Visualizing the relationships between descriptors and activity using scatter plots.
- Analyzing the coefficients and statistical significance of the model parameters.
II. Types of QSAR Models
Linear and non-linear QSAR models are two main approaches used to establish quantitative relationships between molecular structure and biological activity.
- Linear QSAR models assume a linear relationship between the molecular descriptors and the biological activity:

where wi are the model coefficients, b is the intercept, and ∑ is the error term.
Examples of linear QSAR models include multiple linear regression (MLR) and partial least squares (PLS).
- Non-linear QSAR models can capture more complex relationships by using non-linear functions, such as artificial neural networks (ANNs) or support vector machines (SVMs). The general form of a non-linear QSAR model is:

where 𝑓 is a non-linear function learned from the data.
The choice between linear and non-linear QSAR models depends on the complexity of the structure-activity relationship and the size and quality of the available data. Non-linear models can capture more complex patterns but require larger datasets for training and are more prone to overfitting. In a comparative study, both linear PLS and non-linear ANN QSAR models were developed for predicting the antioxidant capacity of phenolic compounds. The non-linear ANN model showed stronger predictive performance, highlighting the importance of non-linear relationships between molecular descriptors and biological activity in this case.
QSAR Workflow
A typical QSAR modeling workflow contains the following key steps:
- Compile a dataset of chemical structures and their associated biological activities or properties. Ensure the dataset is of high quality and representative of the chemical space of interest.
- Calculate a diverse set of molecular descriptors that capture the structural, physicochemical, and electronic properties of the compounds.
- Select the most relevant descriptors using feature selection techniques to avoid overfitting and improve model interpretability.
- Split the dataset into training and test sets, often using methods like the Kennard-Stone algorithm, to enable proper model validation.
- Build QSAR models using regression or classification algorithms such as multiple linear regression (MLR), partial least squares (PLS), or random forest.
- Validate the models using internal (e.g. cross-validation) and external test sets to assess their predictive performance and robustness.
- Evaluate the applicability domain of the models to determine the chemical space where the models can make reliable predictions.

Chemical Descriptors for QSAR
Chemical descriptors are numerical representations of the structural, physicochemical, and electronic properties of molecules. They play a fundamental role in QSAR modeling by providing a quantitative way to encode the chemical information of molecules
Types of Descriptors for QSAR
Despite significant advancements in drug design, defining the molecular structure of biologically active compounds through descriptors remains the primary approach for identifying new lead molecules. Descriptors, which quantify a molecule's chemical characteristics in numerical form, are of different types - constitutional, topological, geometric, thermodynamic and electronic.
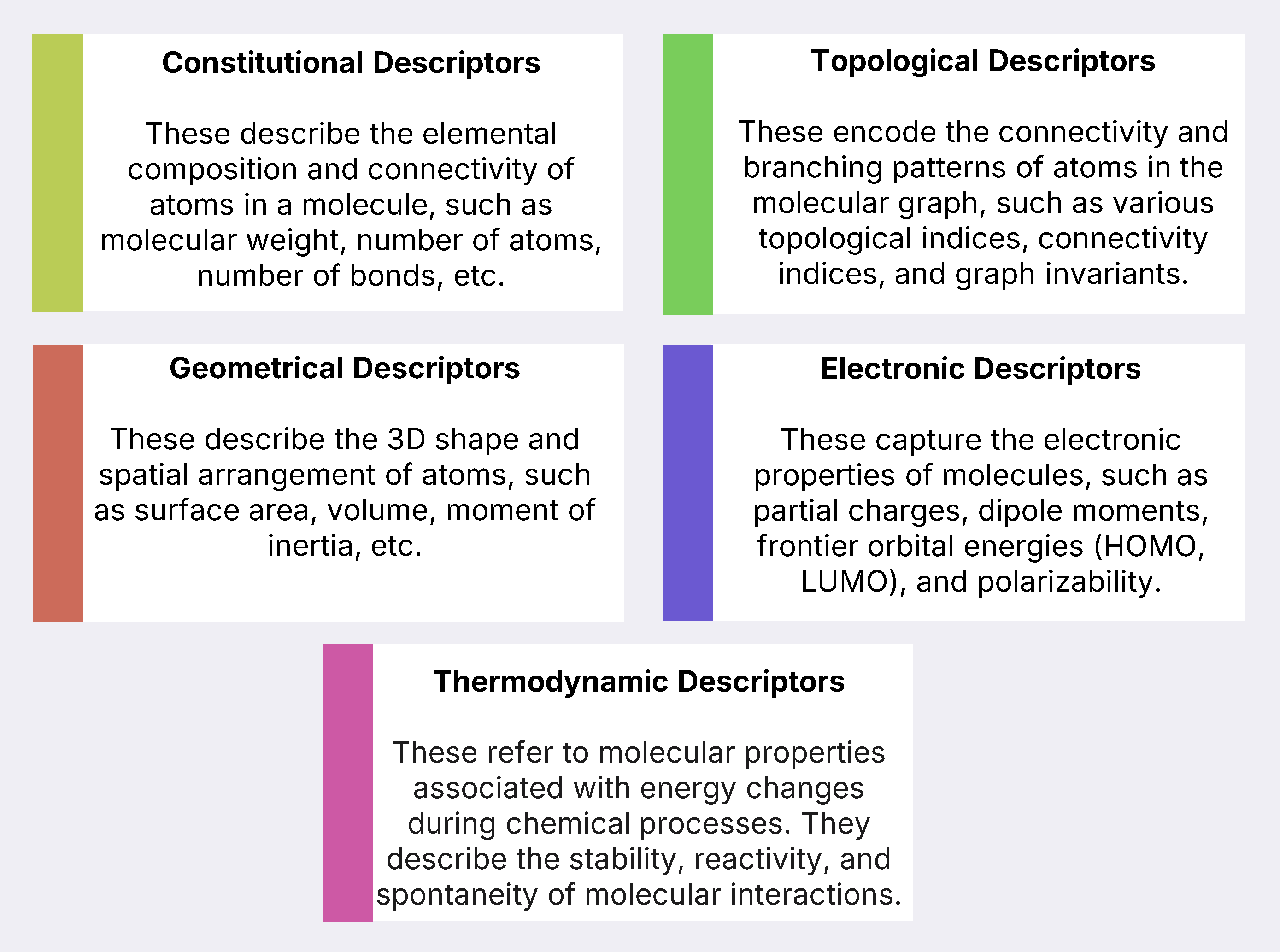
Descriptor Calculation and Software Tools for QSAR
Numerous software packages are available to calculate a wide variety of molecular descriptors, including:
- PaDEL-Descriptor
- Dragon
- RDKit
- Mordred
- ChemAxon
- OpenBabel
These tools can generate hundreds to thousands of descriptors for a given set of molecules. Careful selection of the most relevant descriptors is crucial to building robust and interpretable QSAR models.
Related Reading: A curated list of top cheminformatics software and libraries.
Data Preparation for QSAR
The quality and curation of the datasets are crucial for developing robust and reliable QSAR models.
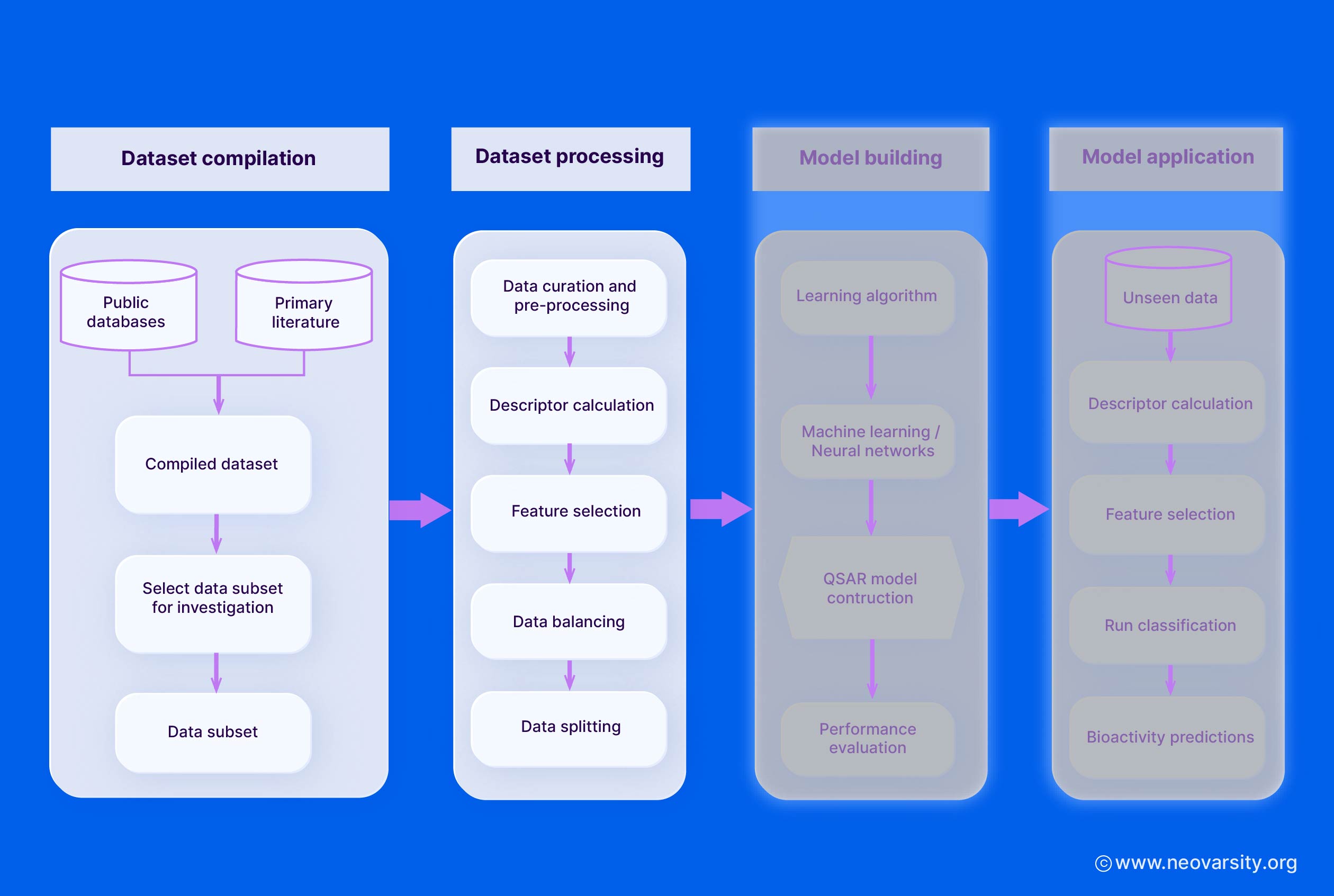
The key steps in data preparation include:
1. Dataset Collection
Compile a dataset of chemical structures and their associated biological activities or properties from reliable sources, such as literature, patents, and public/private databases.
Ensure the dataset covers a diverse chemical space relevant to the problem at hand.
Carefully document the data sources, experimental conditions, and any other metadata.
2. Data Cleaning and Preprocessing
- Remove any duplicate, ambiguous, or erroneous data entries.
- Standardize the chemical structures (e.g., remove salts, normalize tautomers, handle stereochemistry).
- Convert all biological activities to a common unit and scale.
- Handle any outliers or extreme values in the data.
3. Handling Missing Values
- Identify the extent and patterns of missing data in the dataset.
- Employ appropriate techniques to handle missing values, such as:
- Removing compounds with missing data (if the fraction of missing data is low).
- Imputing missing values using methods like k-nearest neighbors, matrix factorization, or QSAR-based prediction.
4. Data Normalization and Scaling
Normalize the biological activity data to a common scale (e.g., log-transform, standardize to z-scores).
Scale the molecular descriptors to have zero mean and unit variance to ensure equal contribution during model training.
Avoid normalization techniques that can introduce bias, such as min-max scaling.
To ensure robust model development and evaluation, the cleaned and preprocessed dataset should be divided into training, validation, and external test sets. The external test set must be reserved exclusively for final model assessment, remaining independent of model tuning and selection.
Related reading: Cheminformatics Data Pipelining Tutorial Part 1: The Foundation.
QSAR Model Building
The model-building stage involves selecting appropriate algorithms, performing feature selection, and validating the models using training and test sets.
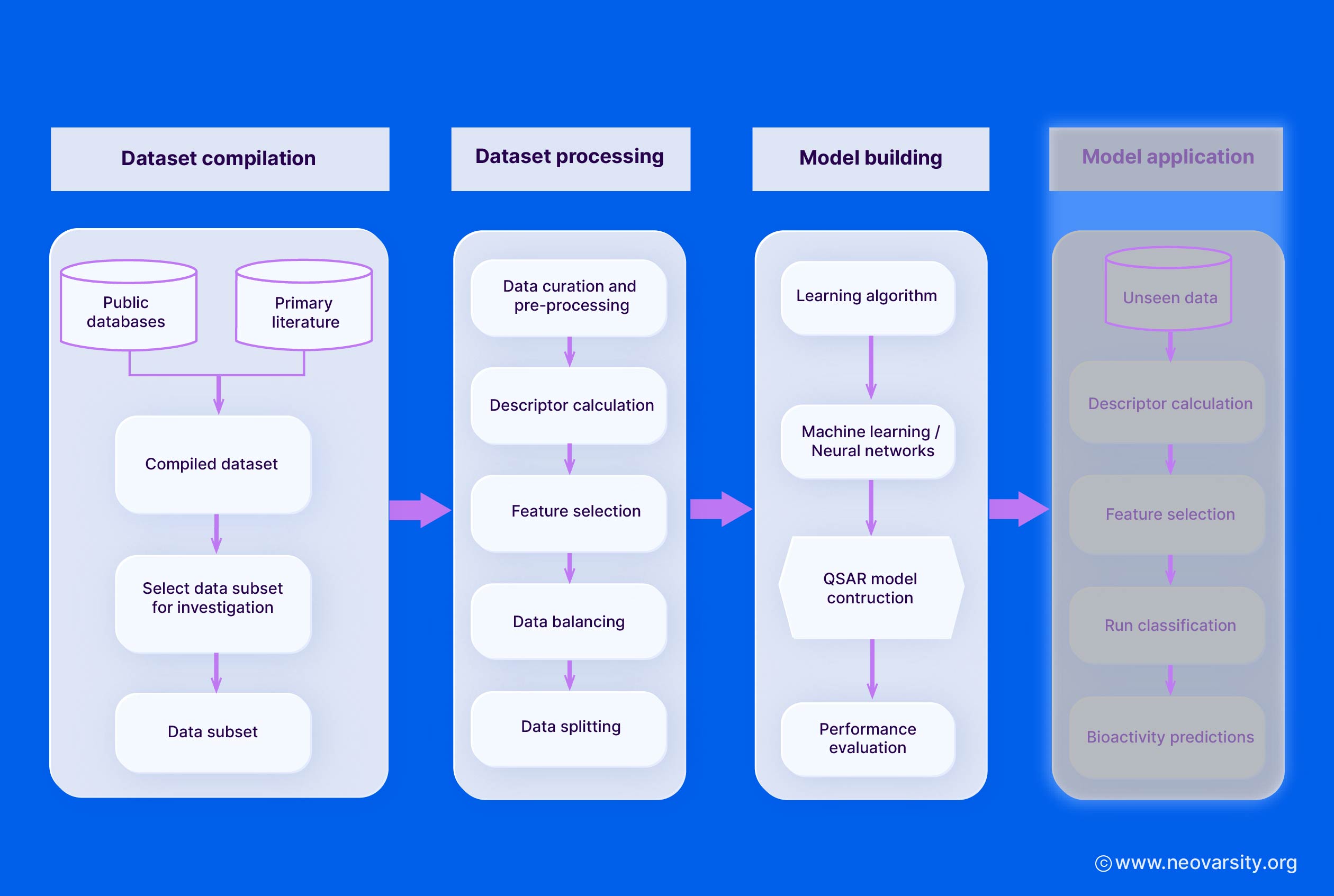
I. Selection of Algorithms
Some commonly used QSAR modeling algorithms include:
- Multiple Linear Regression (MLR): A simple and interpretable linear model that relates the molecular descriptors to the biological activity.
- Partial Least Squares (PLS): A regression technique that handles multicollinearity in the descriptor data and can deal with a large number of descriptors.
- Support Vector Machines (SVM): A non-linear modeling approach that can capture complex structure-activity relationships and is robust to overfitting.
- Neural Networks (NN): Flexible non-linear models that can learn intricate patterns in the data, but may require larger datasets and are less interpretable.
The choice of algorithm depends on the complexity of the structure-activity relationship, the size and quality of the dataset, and the desired level of model interpretability.
II. Feature Selection Methods
Feature selection is crucial to identify the most relevant molecular descriptors and improve the model's predictive performance and interpretability.
Common feature selection methods include:
Filter Methods: Rank descriptors based on their individual correlation or statistical significance (e.g., correlation coefficient, t-test, ANOVA).
Wrapper Methods: Use the modeling algorithm itself to evaluate different subsets of descriptors and select the most informative ones (e.g., genetic algorithms, simulated annealing).
Embedded Methods: Perform feature selection as part of the model training process (e.g., LASSO regression, random forest feature importance).
III. Training and Validation Sets
The dataset is typically split into training, validation, and external test sets:
- The training set is used to build the QSAR models.
- The validation set is used to tune model hyperparameters and select the final model.
- The external test set is used to assess the model's predictive performance on unseen data.
IV. Cross-validation Techniques
Cross-validation is used to estimate the model's predictive performance during the training process. Common techniques include:
k-fold cross-validation: Divide the training set into k subsets, train on k-1 subsets and test on the remaining subset, repeating this process k times.
Leave-one-out cross-validation: Use a single compound as the test set and the remaining compounds as the training set, repeating this for all compounds.
Cross-validation helps prevent overfitting and provides a more reliable estimate of the model's generalization ability.
QSAR Model Validation
Model validation is a critical step in the QSAR modeling workflow to assess the predictive performance, robustness, and reliability of the developed models. It involves both internal and external validation techniques.
Internal Validation
Internal validation methods use the training data to estimate the model's predictive performance. Common techniques include:
1. Cross-Validation (CV):
- Divide the training set into k subsets (folds).
- Train the model on k-1 folds and test on the remaining fold.
- Repeat this process k times, using each fold as the test set once.
- Calculate the average performance across all folds.
2. Leave-One-Out (LOO) CV:
- A special case of CV where k equals the number of compounds in the training set.
- Train the model on all but one compound and test on the left-out compound. Repeat this process for each compound in the training set.
Internal validation provides an estimate of the model's predictive performance on new data but may be optimistic due to the use of the same data for training and validation.
External Validation
External validation uses an independent test set that was not used during model development to assess the model's predictive performance on unseen data. This provides a more realistic estimate of the model's performance in real-world applications. Techniques include:
1. Test Set Validation:
- Split the dataset into training and test sets.
- Develop the model using the training set.
- Evaluate the model's performance on the test set.
2. y-Randomization:
- Randomly shuffle the activity values (y-values) in the training set.
- Build a model using the shuffled data.
- Evaluate the model's performance on the test set.
- Repeat this process multiple times.
- If the model performs well on the randomized data, it may be overfitting.
Statistical Metrics
Various statistical metrics are used to quantify the model's predictive performance:

For classification tasks, additional metrics include:
Accuracy: The proportion of true results (both true positives and true negatives) among the total number of cases examined.
Precision: The proportion of true positive results in all positive predictions.
Recall (Sensitivity): The proportion of true positive results in all actual positive cases.
Specificity: The proportion of true negative results in all actual negative cases.
F1 Score: The harmonic mean of precision and recall provides a balance between the two.
MCC (Matthews Correlation Coefficient): A balanced measure that takes into account true and false positives and negatives, suitable for imbalanced datasets.

The choice of metrics depends on the type of activity (continuous or categorical) and the specific requirements of the application. Rigorous validation using both internal and external techniques is essential to ensure the developed QSAR models are reliable and robust, and can be confidently applied to virtual screening and lead optimization tasks.
Interpretation of QSAR Models
Interpreting QSAR models is crucial to understanding the complex relationships between molecular structure and biological activity and to guide the design of new, more potent compounds.
Some key techniques for QSAR model interpretation include:
1. Understanding Model Coefficients
For linear QSAR models, such as multiple linear regression (MLR) or partial least squares (PLS), the model coefficients can provide insights into the influence of individual molecular descriptors on the predicted activity:
The magnitude of a coefficient indicates the relative importance of the corresponding descriptor.
The sign of the coefficient (positive or negative) suggests whether the descriptor is positively or negatively correlated with the activity.
Statistical significance tests can identify the descriptors that have a statistically significant impact on the model.
2. Analyzing Descriptor Contributions
For both linear and non-linear QSAR models, the contributions of individual descriptors can be analyzed:
Feature importance methods, such as permutation importance or Shapley values, can quantify the relative importance of each descriptor.
Partial dependence plots can visualize the relationship between a descriptor and the predicted activity while holding other descriptors constant.
Sensitivity analysis can assess how changes in a descriptor value affect the model output.
3. Visualization Techniques
Visualizing the QSAR models and the underlying structure-activity relationships can provide valuable insights:
- Scatter plots of predicted vs. observed activities can reveal systematic errors or outliers.
- Heatmaps can highlight the regions of the molecule that contribute most to the predicted activity.
- Three-dimensional pharmacophore models can identify the key structural features and their spatial arrangements that are important for biological activity.
Common Challenges in QSAR Modeling
QSAR modeling, like any data-driven approach, faces several challenges that need to be addressed to ensure the reliability and robustness of the developed models. Some of the common challenges include:
1. Overfitting and Underfitting
Overfitting occurs when a model learns the training data too well, capturing noise and irrelevant patterns, which leads to poor generalization to new, unseen data. This typically happens when the model is overly complex, such as having too many parameters relative to the training dataset size. On the other hand, underfitting arises when a model is too simple to capture the underlying structure-activity relationship, resulting in poor performance on both the training and test data due to an inability to learn meaningful patterns.
Strategies to address overfitting and underfitting include:
- Careful feature selection to identify the most relevant descriptors
- Regularization techniques (e.g., L1/L2 regularization, dropout) to control model complexity
- Rigorous model validation using internal and external test sets
2. Descriptor Redundancy
QSAR models often include a large number of molecular descriptors, many of which may be correlated or redundant, leading to overfitting, instability, and difficulty in model interpretation.
Techniques to handle descriptor redundancy include:
- Feature selection methods to identify the most informative and non-redundant descriptors
- Principal component analysis (PCA) or other dimensionality reduction techniques to transform the descriptor space
3. Predictive Uncertainty
QSAR models provide point estimates of the predicted activities but do not quantify the uncertainty associated with these predictions. Estimating the predictive uncertainty is crucial for making informed decisions, especially in drug discovery applications where the consequences of false predictions can be high.
Approaches to address predictive uncertainty include:
- Bayesian modeling techniques that provide probabilistic predictions and credible intervals.
- Ensemble methods that combine multiple models to estimate the prediction variance.
- Applicability domain analysis to determine the chemical space where the model can make reliable predictions.
Advanced QSAR Techniques
QSAR modeling has evolved beyond the traditional linear and non-linear regression approaches to include more sophisticated techniques that can capture complex structure-activity relationships. Some of the advanced QSAR methods include:
3D-QSAR
3D-QSAR methods incorporate the three-dimensional (3D) structural information of molecules to build predictive models. Two prominent 3D-QSAR techniques are:
1. Comparative Molecular Field Analysis (CoMFA)
CoMFA represents the 3D molecular structure using steric and electrostatic fields, enabling a detailed analysis of molecular interactions. It aligns molecules based on a common structural framework for consistent comparison. Finally, CoMFA builds a regression model that correlates 3D field descriptors with biological activity, providing predictive insights into structure-activity relationships.
2. Comparative Molecular Similarity Indices Analysis (CoMSIA)
CoMSIA is similar to CoMFA but incorporates additional physicochemical descriptors, such as hydrogen bonding, hydrophobicity, and partial charges. This approach provides more comprehensive information on 3D structure-activity relationships, enhancing the predictive power of the model.
3D-QSAR models can provide valuable insights into the structural features and interactions that govern biological activity, guiding the design of new, more potent compounds.
Machine Learning Approaches in QSAR
Advanced machine learning algorithms have been widely adopted in QSAR modeling due to their ability to capture complex non-linear relationships. Some frequently used algorithms are:
Random Forests (RF)
It is an ensemble learning method that combines multiple decision trees to improve predictive accuracy and robustness. It is resistant to overfitting and effectively handles high-dimensional descriptor spaces. Additionally, RF provides feature importance measures, helping identify key structural determinants in molecular modeling.
Gradient Boosting Machines (GBM)
It is an ensemble technique that iteratively builds weak prediction models and combines them to enhance overall performance. It can handle both continuous and categorical endpoints, making it highly versatile. Additionally, GBM offers flexible feature engineering and automatically selects the most relevant descriptors for improved predictive accuracy.
These machine-learning techniques have shown superior predictive performance compared to traditional linear and non-linear QSAR models, especially for large and diverse datasets.
Deep Learning in QSAR
Recent advancements in deep learning have significantly enhanced QSAR modeling by enabling automatic feature extraction and pattern recognition.
For example, neural networks can learn relevant features directly from raw molecular representations, such as SMILES and molecular graphs, without requiring manual descriptor calculation.
Convolutional neural networks (CNNs) can capture local structural patterns in molecules, similar to how they detect features in image recognition, making them useful for identifying spatial relationships in chemical structures.
Recurrent neural networks (RNNs) are particularly effective for handling sequential molecular representations like SMILES strings, allowing for improved modeling of molecular sequences.
Graph neural networks (GNNs) go a step further. They can directly operate on molecular graph structures, preserving the inherent connectivity information between atoms and bonds. This allows them to capture complex molecular relationships, making them highly effective for QSAR modeling and drug discovery applications.
Related reading: 11 reasons to master cheminformatics before machine learning in drug discovery
QSAR Software and Tools
Popular QSAR Software and Tools
QSAR modeling relies on a variety of software tools and platforms to facilitate the different steps of the workflow, from data preparation to model building and validation. Here is an overview of some of the popular QSAR software and tools:

Open Source vs Commercial QSAR Tools
Open-source tools like QSAR Toolbox and KNIME offer free access and flexibility but may require more technical expertise to set up and use. Commercial tools like Dragon and MOE provide more comprehensive features, user support, and a polished interface, but require licensing fees. The choice between open-source and commercial tools depends on the specific needs of the project, available resources, and the level of expertise of the research team.
Want to learn QSAR modeling through coding? Explore the Cheminformatics: Tools and Applications course now!
Conclusion
In conclusion, QSAR modeling is a powerful computational technique that enables the quantitative prediction of biological activities and properties of molecules based on their chemical structure. In this comprehensive beginner's guide to QSAR modeling in cheminformatics, we have covered the key concepts, workflow, and applications of QSAR modeling applied to the biopharma industry.
Here is a summary of the key points:
QSAR models represent molecules as numerical vectors of molecular descriptors and aim to establish a quantitative relationship between these descriptors and the biological activity of interest.
The QSAR modeling workflow involves data preparation, descriptor calculation, feature selection, model building, and rigorous validation using internal and external test sets.
Various linear and non-linear modeling algorithms, such as MLR, PLS, SVM, and neural networks, are used in QSAR depending on the complexity of the structure-activity relationship and the available data.
Interpreting QSAR models is crucial to understanding the key structural features driving biological activity and guiding the design of new, more potent compounds.
QSAR modeling faces challenges such as overfitting, descriptor redundancy, and predictive uncertainty, which need to be addressed through careful model development and validation.
Advanced QSAR techniques, such as 3D-QSAR, machine learning, and deep learning, have shown promising results in capturing complex structure-activity relationships.
A wide range of open-source and commercial software tools are available to facilitate different steps of the QSAR workflow.
Future Outlook
The future of QSAR lies in its integration with other computational and experimental methods, leveraging big data and machine learning, and enabling personalized medicine. Combining QSAR with molecular docking, systems biology, and real-world evidence can provide a more comprehensive understanding of structure-activity relationships.
Applying scalable QSAR algorithms to large, diverse datasets and incorporating uncertainty quantification in predictions will be crucial for realizing the full potential of QSAR in drug discovery and beyond. Undoubtedly, QSAR modeling has become an indispensable tool in the arsenal of computational chemists and biologists, enabling data-driven decision-making in various domains, from drug discovery to chemical safety assessment.
As the field evolves, particularly with the integration of AI and machine learning, QSAR will become increasingly vital in accelerating the development of safer, more effective, and innovative chemicals and drugs.