How Machine Learning Models Learn Chemistry from SMILES
Learn how to format, tokenize, and prepare SMILES so machine learning models can process, embed, and generate molecules.
10 min read
May 8th, 2025
Last updated: May 10th, 2025


Introduction
Before a machine can predict whether a molecule is toxic, bioactive, or synthesizable, it first needs to understand what the molecule is.
That may sound simple, but it’s not, because molecules aren’t numbers, images, or plain text.
They are complex topological, spatial, and chemical structures that must be translated into a machine-readable format. For most modern machine learning workflows, that format often starts with SMILES.
In this post, we’ll explore how SMILES is used in machine learning: how it’s structured, how it’s tokenized, and how models learn to process, embed, and generate it.
This blog is the second part of an eight part series which dives deep into how machine learning reads chemical structures. Check out the first part here.
What is SMILES?
SMILES, or the Simplified Molecular Input Line Entry System, is a way to represent a molecule as a compact ASCII string. It encodes atoms, bonds, branching, ring closures, and stereochemistry in a linear format that's easy for machines to parse and store.
While simple to write, SMILES encodes rich structural information. So understanding its rules and how to use them is essential for building effective machine learning models.
Let’s quickly recap the fundamentals of SMILES format.
Basic syntax rules of SMILES
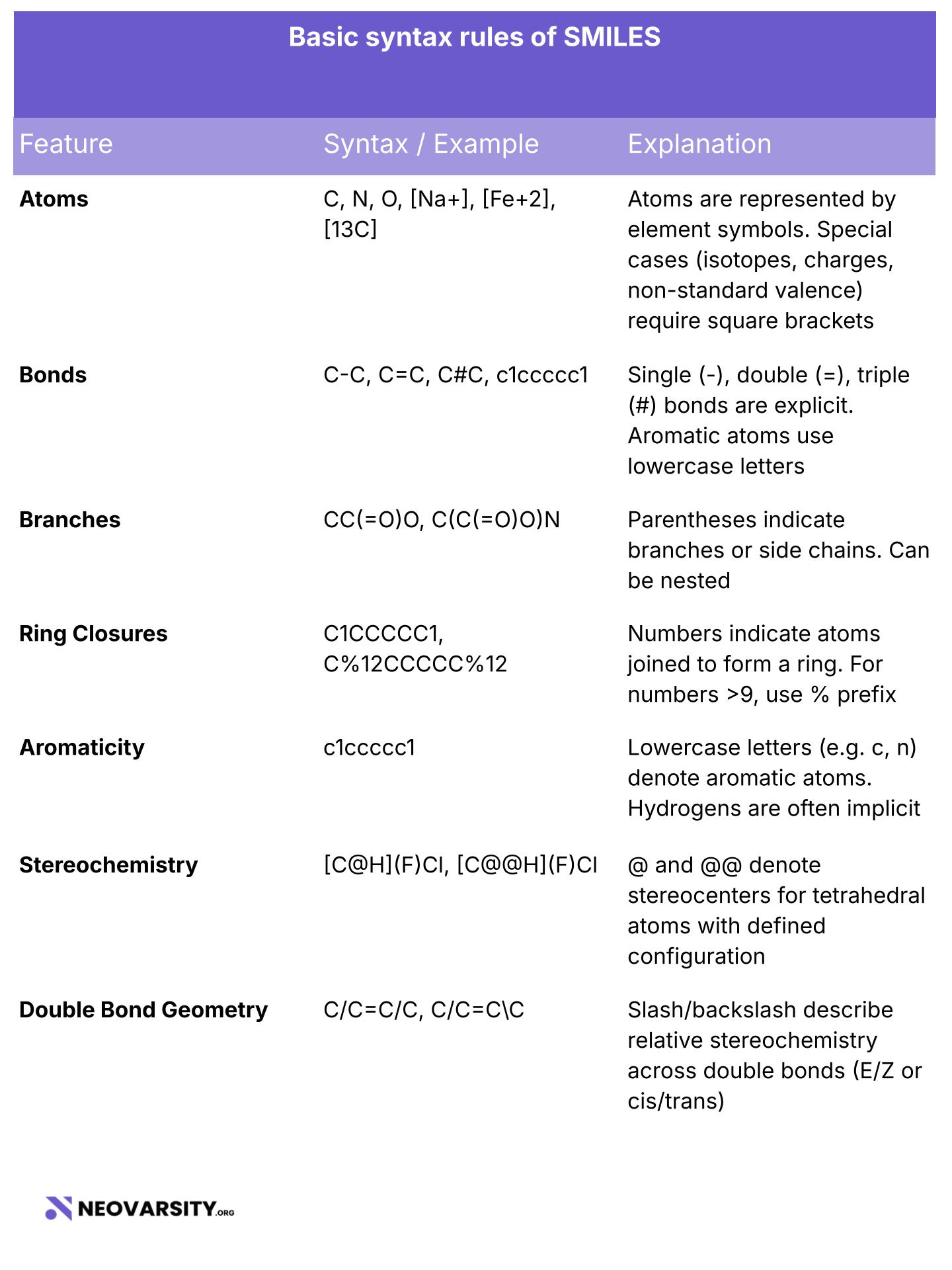
Atoms are represented by their atomic symbols such as C, N, O, P, etc. Atoms with charges, isotopes, or unusual valence must be enclosed in square brackets, for example: [Na+], [Fe+2], [13C].
Bonds are either implied (C-C) or explicitly represented using symbols: single bonds as -, double bonds as =, triple bonds as #, and aromatic bonds using lowercase atoms, as in c1ccccc1 for benzene.
Branches are enclosed in parentheses, as shown in CC(=O)O for acetic acid. Nested branches are allowed, for example: C(C(=O)O)N.
Ring closures are denoted using numbers, such as C1CCCCC1 for cyclohexane. For ring labels beyond 9, double digits are used with a percent sign, like %10 or %11.
Aromaticity is indicated using lowercase atom symbols, for example: c1ccccc1. Hydrogens on aromatic rings are assumed to be implicit.
Stereochemistry is indicated using @ and @@ for chiral centers, as in [C@H] and [C@@H].
Double bond geometry is represented using / and \ to specify E/Z isomerism.
Let’s take the example of the classic molecule aspirin.
Below is its SMILES representation along with its chemical breakdown.
CC(=O)OC1=CC=CC=C1C(=O)O
In this structure, CC(=O)O represents the acetyl group, which is an ethyl chain attached to a ketone and an oxygen atom. C1=CC=CC=C1 represents the aromatic ring, and the final C(=O)O corresponds to the carboxylic acid group. The ring is denoted by repeating the label 1, which indicates the start and end points of the cycle.
At the same time, it’s important to remember that SMILES can also represent invalid structures, and understanding this is crucial.
For instance, the popular open-source cheminformatics software RDKit and others will fail to parse, or produce incorrect structures.
This is why data curation is critical before using SMILES in model training.
Here are a few quick examples.
CC1CC1C1 # Ambiguous ring label reuse C(C(C # Unmatched parentheses CN=C(C) # Bond without second atom
While there are specialized steps to verify and potentially correct SMILES strings, the easiest way is to use RDKit to visualize them and check if they match the expected chemical structure.
Below is the code syntax to do this:
from rdkit import Chem
from rdkit.Chem import Draw
mol = Chem.MolFromSmiles("CC(=O)OC1=CC=CC=C1C(=O)O")
Draw.MolToImage(mol)
This approach is especially useful when we are working with just a handful of examples.
If you want to dive deeper, the Cheminformatics: Tools and Applications Course covers this in detail, including how to validate and correct SMILES across large datasets.
Why rules matter for machine learning?
When building tokenizers, data loaders, or generative models, a deep understanding of these rules is essential.
A missed bracket or misused bond symbol in SMILES might render a molecule invalid or worse, syntactically valid but chemically nonsensical.
Knowing these rules also helps in several ways, such as building synthetic SMILES generators that consistently create valid strings, applying grammar-based constraints in models like VAEs or Transformers, and designing preprocessing steps that can catch malformed or corrupted data before it affects model performance.
Why we use SMILES for molecular machine learning?
The simple reason we use SMILES in molecular machine learning is that it offers a compact, one-dimensional sequence that is ideal for feeding into models like RNNs or Transformers.
When parsed correctly, SMILES allows for the lossless reconstruction of molecular structures. It also provides a strong foundation for string-based generation, much like how language models handle sentences.
However, it’s important to recognize that SMILES come with their own set of challenges.
One key issue we should be aware of is that SMILES is not unique. Multiple non-canonical forms can represent the same molecule.
This means two different SMILES strings might describe the same structure. We also need to be careful with syntax, as even small mistakes can lead to invalid or entirely different molecules.
Another major limitation is that SMILES strings lack spatial information, which can be critical for accurately understanding molecular behavior.
As we start building generative models or training classifiers, it is very important to keep these challenges in mind and it will help us avoid common pitfalls and create stronger, more reliable systems.
Turning SMILES into model-ready inputs
Before a machine learning model can start learning from SMILES strings, the raw text must be processed into a form it can actually work with. This brings us to an important step: Tokenization.
Machine learning models cannot understand raw characters directly. They need discrete tokens that map to meaningful chemical information.
Without this step, the model would simply treat SMILES as a random jumble of letters and symbols, completely missing the underlying chemistry.
A common mistake is using a naive, character-level tokenizer.
This approach breaks the SMILES string down letter by letter, which fails when dealing with multi-character atoms like "Cl" (chlorine) or "Br" (bromine), or more complex bracketed species like "[Na+]".
If not handled properly, the model would misinterpret these critical structures.
To avoid this, regex-based tokenizers are the standard.
They allow us to correctly split SMILES strings into chemically meaningful units, preserving the structure and ensuring the model learns from accurate representations.
In the next part of this blog series on 'How machine learning reads chemical structures', we will dive into how these tokenizers are built and what best practices you should follow to prepare your data correctly.
But for now, let’s focus on understanding a standard regex tokenizer for SMILES.
While the field of handling SMILES for machine learning models has advanced significantly with many automated tools (also covered in the Cheminformatics: Tools and Applications course), it is still worth taking the time to learn the fundamentals.
This is especially important if you are in the early stages of building your skills.
import re
def tokenize_smiles(smiles):
pattern = r"(\[[^\[\]]{1,6}\])"
tokens = re.split(pattern, smiles)
result = []
for token in tokens:
if token.startswith('['):
result.append(token)
else:
result.extend(list(token))
return result
# Example usage
print(tokenize_smiles('CC(=O)OC1=CC=CC=C1C(=O)O'))
Output:
['C', 'C', '(', '=', 'O', ')', 'O', 'C', '1', '=', 'C', 'C', '=', 'C', 'C', '=', 'C', '1', 'C', '(', '=', 'O', ')', 'O']
These tokens form the basis for vocabulary-building and embedding.
Turning SMILES tokens into embeddings
Once you tokenize a SMILES string, the next step is to convert each token into a numerical format that an machine learning model can understand.
This process, called embedding, is similar to how words are embedded in natural language processing tasks.
Each SMILES token is mapped to a vector that captures its meaning and relationship to other tokens.
There are several techniques for embedding SMILES, depending on the complexity and goals of your model. One basic method is one-hot encoding, where each token is represented by a binary vector.
While easy to implement, this approach does not capture any relationships between tokens. A more powerful method involves learned embeddings, where models like LSTMs and GRUs develop dense vector representations during training, allowing the model to better pick up chemical patterns.
In transformer-based architectures, positional embeddings are often added to provide information about the sequence order, which is crucial because SMILES strings are highly order-sensitive.
Finally, pretrained embeddings from models like ChemBERTa and MolBERT offer a rich chemical context right from the start, as they have been trained on large chemical datasets.
Choosing the right embedding approach is an important decision that can significantly influence how well your model learns and generalizes from molecular data.
Here’s a quick example of how to implement embeddings using the machine learning library PyTorch.
It’s also worth mentioning that embeddings are often at the core of building SMILES-based machine learning models.
You can dive deeper into these concepts in the following curriculums: Molecular Machine Learning Foundation, Advanced Machine Learning for Drug Discovery, and Generative AI for Small Molecule Drug Discovery.
import torch.nn as nn vocab_size = 64 embedding_dim = 256 embedding_layer = nn.Embedding(vocab_size, embedding_dim)
These embeddings are then fed into sequence models like RNNs or Transformers.
Which machine learning models learn from SMILES?
Once SMILES strings are tokenized and embedded, the next step is choosing a machine learning architecture that can effectively learn from them.
Two main types of models are commonly used for this purpose: recurrent models and transformer models.
Recurrent models, such as LSTMs and GRUs, process SMILES as sequences, treating the string similarly to how natural language is handled.
These models are useful for tasks like property prediction and molecule generation, where understanding the sequential flow of the SMILES string is important.
However, recurrent models can struggle with longer molecules, as they are prone to vanishing gradients, which can limit their ability to capture long-range dependencies.
Transformer models offer a powerful alternative. Using self-attention mechanisms, transformers can learn long-range dependencies much more effectively than recurrent networks.
They are widely used in generative modeling, including GPT-style molecule generation.
Some notable examples include ChemBERTa, designed for chemical property prediction, and the Molecular Transformer, which is specifically used for reaction prediction tasks.
Choosing between these architectures depends on the specific task and dataset, but both have become foundational tools in SMILES-based machine learning workflows.
Tokenized SMILES → Embedding Layer → Transformer Encoder → Output Layer (classification or regression)
How to fix SMILES issues for molecular machine learning?
Because SMILES strings are not unique, a single molecule can have multiple valid SMILES representations.
This variability, often referred to as SMILES fragility, is a significant challenge in molecular machine learning. However, if done right, this variability can actually be used to our advantage.
One effective approach is SMILES enumeration, which is a simple technique that generates different valid SMILES strings for the same molecule.
It can also be used to improve model robustness through data augmentation. For example, using RDKit, we can generate randomized SMILES by applying slight changes in atom ordering while preserving the molecule's structure.
Here’s a quick example on how to implement it:
from rdkit import Chem mol = Chem.MolFromSmiles(smiles) print([Chem.MolToSmiles(mol, doRandom=True) for _ in range(5)])
Another way to tackle SMILES fragility is by using SELFIES (Self-Referencing Embedded Strings), which basically offers a more robust molecular representation than SMILES.
In practicality, SELFIES act as a drop-in replacement for SMILES and guarantee that any sequence produced can be decoded into a valid molecule.
Here’s a quick example:
import selfies as sf
s = sf.encoder(smiles)
print("SELFIES:", s)
print("Decoded:", sf.decoder(s))
Using SELFIES can significantly reduce invalid generations in generative models, making it a valuable tool when building more reliable molecular design pipelines. Here is the GitHub repository for SELFIES. Have a look!
What we can build with SMILES in machine learning?
When working with SMILES, we open up a wide range of possibilities for applying machine learning to drug discovery and chemistry.
We can start with QSAR modeling, using SMILES to predict important properties like toxicity, solubility, and binding affinity.
We can also explore molecular generation, where models like VAEs, GANs, or Transformers create entirely new molecules from learned patterns.
For those of us interested in synthesis planning, SMILES strings can serve as both input and output in retrosynthesis prediction, allowing sequence-to-sequence models to map reactants to products.
We can even use SMILES for property optimization, applying latent vector manipulations or reinforcement learning techniques to design better-performing molecules.
But across all of these applications, our success will depend on how well we handle tokenization, create clean embeddings, and choose the right model architectures to capture chemical complexity.
Stay tuned for the next part of this series that discusses exactly that!
Final words
Now that we’ve explored what SMILES are and how they’re used in machine learning for drug discovery, it’s clear that SMILES serve as the gateway for molecules to enter the machine learning pipeline.
They enable powerful models to classify, generate, and optimize compounds but only when the syntax, structure, and semantics are handled correctly.
So it can be confidently said that handling SMILES is foundational for molecular machine learning.
Of course, SMILES come with their own set of challenges. One of the biggest issues is non-uniqueness, a single molecule can have many different SMILES representations.
There’s also the problem of stereochemistry, which is often omitted or difficult to encode precisely.
We face data imbalance as well, with bioactive compounds being far less common than inactive ones. And in low-data, high-dimensional settings, overfitting becomes a real concern.
That’s why understanding how to tokenize, embed, and process SMILES is essential for anyone building machine learning systems in cheminformatics or drug discovery.
In the next part of the series, we’ll take a deeper look at tokenization: how to design vocabularies, handle special tokens, and prepare SMILES for transformer-based large-scale modeling.
Learn by doing!
Join Neovarsity's certified cheminformatics course.
Get hands-on experience with SMILES-based modeling using tools like RDKit, PyTorch, and SELFIES.