How Machine Learning Reads Chemical Structures
In this series, we're taking a deep technical dive into how chemical structures are read, represented, and interpreted in molecular machine learning.
4 min read
May 8th, 2025
Last updated: May 10th, 2025


Introduction
In molecular machine learning, whether we're working in drug discovery, material science, crop science, or any related field, data representation is foundational.
Molecules are inherently complex. They exist as high-dimensional structures with rich chemical behavior.
For a machine learning system to learn from them, it first needs to read and interpret them.
This means we have to choose the right way to encode these complex molecular structures.
So, before we build a model to predict solubility, generate a novel compound, or suggest a synthetic route, we first need to translate the molecule into a form the machine can understand.
We need to keep in mind that this translation isn't trivial. The format we choose directly influences what the model can learn and how well it performs.
About this series
In this series, we're going to take a deep technical dive into how chemical structures are represented, interpreted and used in molecular machine learning.
This post is Part I of the series How Machine Learning Reads Chemical Structures. In this series, we will cover:
Part III: Tokenizing Chemical Structures
Part IV: Embedding Molecules
Part V: Graph-Based Representations
Part VI: Molecular Generation Models
Part VII: Robust Representations and Augmentation
Part VIII: Hybrid and 3D Molecular Inputs
We’ll cover some basics, but we’ll go much deeper on the technical aspects, focusing on those of us already working on or interested in tasks like QSAR modeling, generative design, retrosynthesis, and molecular property prediction, etc.
If you’re just starting out, especially in cheminformatics and molecular machine learning, we recommend going through these guides first:
11 reasons to master cheminformatics before machine learning in drug discovery
Cheminformatics: an in-depth guide for beginners
A curated list of cheminformatics software and libraries
A beginner's guide to QSAR modeling in cheminformatics for biopharma
What are molecular representations in machine learning?
Molecular representations are methods for translating the complex 3D structure of molecules, including atoms, bonds, and spatial arrangements, into structured formats that machine learning models can process.
These representations may be numerical, symbolic, or graphical, and are essential for enabling models to learn chemical properties and relationships effectively.
Before we move forward, here’s a quick summary of some of the most commonly used representations.
Main types of molecular representations
| Format | Type | Captures | Ideal for | Limitations |
|---|---|---|---|---|
| SMILES | 1D String | Atoms, bonds, rings | QSAR, sequence models, generative models | Non-unique, syntax-sensitive |
| InChI | 1D String | Canonical connectivity | Search, deduplication | Hard to use in generative workflows |
| Graph | 2D Topology | Atom/bond relationships | Graph neural networks, property prediction | Loses 3D info unless extended |
| 3D Coordinates | 3D Spatial | Atom positions, molecular shape | Docking, conformer generation, QM tasks | Expensive to compute, may not generalize |
| Fingerprints | Bit Vector | Substructure presence/absence | Similarity search, classification | Fixed size, loses structural context |
| Embeddings | Learned Vectors | Latent chemical/structural features | Property prediction, generative design | Data-hungry, lacks interpretability |
Each format reveals different layers of chemical meaning. Some prioritize simplicity, while others focus on precision or generative potential. Choosing the wrong format can hurt model performance or even lead to invalid results.
If you want to dive deeper, you can also check out more details in our Beginner's Guide to Handling Chemical Files and Their Conversion.
Why is molecular representation a challenge in machine learning?
As we work with molecules in machine learning, we quickly realize they are anything but simple.
Molecules exist in three-dimensional space, with atoms connected by bonds and arranged in specific spatial patterns.
Translating this complexity into a format that a model can understand is not straightforward.
No single representation captures every aspect of a molecule’s structure and behavior.
Depending on what we are trying to do, we need to emphasize different features of the molecule.
For property prediction, atomic connectivity might matter most but for docking or conformer analysis, 3D geometry becomes critical.
This is why it's fair to say that molecular representation is a multi-format challenge, and no single format works for every task.
Instead, we rely on a diverse set of representations, each designed and optimized for specific tasks, model types, and practical constraints.
Why representation choice matters in molecular machine learning?
In molecular machine learning, the models we build are only as good as the structures they see.
The way we represent molecules determines which chemical signals the model can learn and which ones it might miss.
Choosing the right format is about balancing trade-offs. We consider accuracy, computational efficiency, and compatibility with our model architecture.
If we want to predict solubility or toxicity, SMILES or 2D descriptors might be enough. If we're designing new molecules, we may need SELFIES or graph-based generators.
If we're modeling binding affinity, 3D geometries become critical. And for retrosynthesis tasks, reaction SMILES and transformation graphs are usually the way to go.
Different tasks demand different representations, and knowing how to choose the right one is key to building models that are both effective and scalable.
Here’s a typical flow of how molecular structures feed into a machine learning model.
SMILES → Tokenizer → Embeddings → ML Model (LSTM/Transformer/GNN) → Output
Each stage adds its own complexity and nuance. How we tokenize and embed a SMILES string can directly influence how well a model generalizes.
The way we structure the model also depends on the type of representation we are working with, whether it is a sequence, a graph, or a spatial format.
In later posts, we will break each of these components down and explore how we can optimize them.
For now, to help you understand the basics more clearly, let’s take a look at the SMILES string for aspirin (acetylsalicylic acid).
The chemical structure can be represented in line notation.
In this case, the SMILES representation is:
CC(=O)OC1=CC=CC=C1C(=O)O
This compact line represents aspirin as a molecule with aromatic rings, ester groups, and carboxylic acids.
But it's just text, models must learn to link this text to chemical meaning.
The same structure can be represented and visualized in different formats.
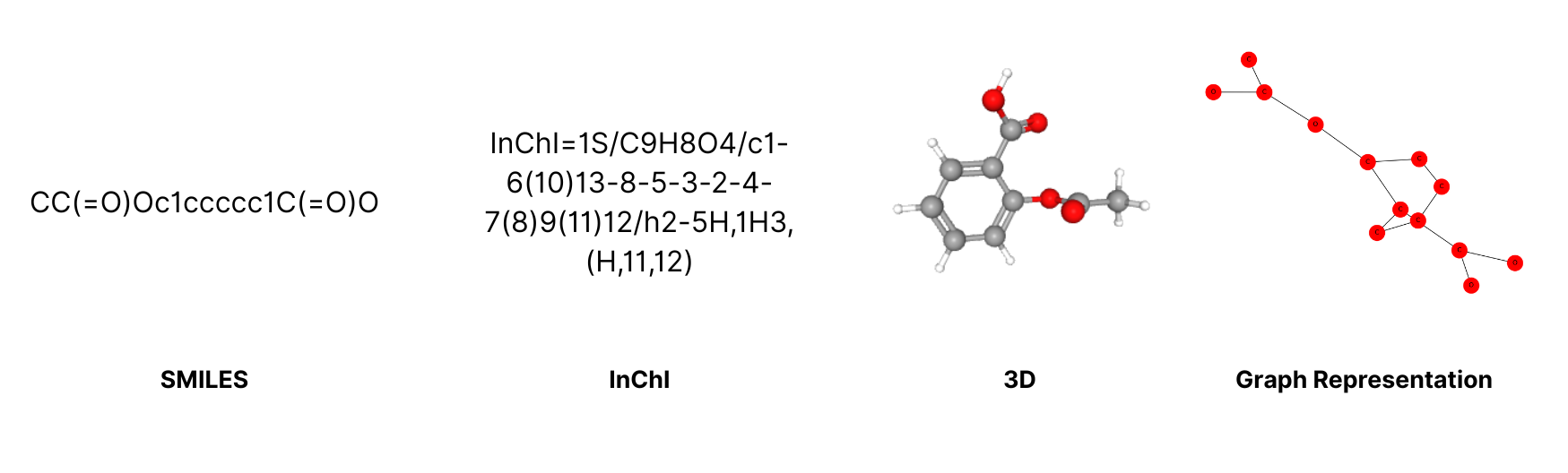
Each of these formats, along with many others, can be used in building machine learning models.
A thorough understanding of them can help us improve model interpretability and design better debugging workflows.
By the way, did you notice that the SMILES string for aspirin shown in the image above and the SMILES written in the text are different? Yet, both are correct!
We will talk more about why that happens in the next part of this series.
What’s next in this series
This is just the start! In the posts that follow, we’ll explore the ML stack that surrounds molecular data.
Topics include:
Tokenizing chemical structures for molecular machine learning
Graph-based modeling with RDKit and PyTorch Geometric
Embedding architectures for molecular sequences
Variational autoencoders and generative models
Robust workflows for dataset curation and augmentation
Hybrid models with spatial or electronic features